

Molecular shredders
The role of read alignment in bioinformatics

There’s a scene in Argo, a Best Picture-winning historical film based on the Iran Hostage Crisis, where US embassy workers attempt to destroy sensitive records by shredding them before the Iranians take over. Unfortunately for the Americans, the Iranians are able to piece back these sensitive documents, uncovering the identities of America’s spies. For the sake of making my cool hook work, imagine that one thing that made things easier for the Iranians was that every single document came embossed with the White House logo, so they just had to recreate the logo in order to align the pieces correctly.

Modern genomics and transcriptomics begin with a process very similar to that shredded paper scene in Argo. In genomics, the “reference genome” is the sequence of A’s, G’s, C’s, and T’s that is most common in humans. The reference genome is like the White House logo. Modern high-throughput sequencers are like the shredder, reading the genome a few dozen base pairs at a time.


So read alignment shouldn’t be that hard, right? Just search through the reference for the matching set of base pairs and then you’re aligned. But of course the reason we bother sequencing genomes is that they are not an exact match with the reference genome. In an actual human, our reads might look like this:

and aligning them would produce:

So to align these reads to the genome we need to do some kind of fuzzy matching, allowing a read to match the reference as long as it’s “close enough.” Luckily, there are a number of open source fuzzy matching algorithms (bowtie, bwa-mem) publicly available that help with this kind of task, as long as you have enough compute to spare.
Only once we have the reads aligned can we perform interesting downstream analyses:
SNP calling
Single Nucleotide Polymorphisms (SNPs, pronounced “snips”) are locations in the genome where we’ve previously observed variation in a single base pair. Where we would normally expect a G, we see an A, T, or C. Because all autosomal genes come in pairs, SNPs need to be “called” to decide if they’re present in one copy of a chromosome, both copies, or neither.

Because position 3 has a “pileup” of G reads, we call position 3 as a heterozygote. Notice that position 5 also has a mismatched read, but we don’t call that position as a heterozygote because we think the T read is due to random noise. Calling SNPs frequently requires this kind of statistical reasoning.
Transcriptomics
Rather than trying to figure out what the deoxyribonucleic acid (DNA) looks like, we can try to figure out how much ribonucleic acid (RNA) it’s producing. RNA modulates the activities of the cell either by itself or by being transcribed into protein, so knowing how much RNA gets produced can help us understand abnormal cell behavior.
RNA transcripts do not map directly to the reference genome, because they usually undergo processing to remove exons, add a 5’ “cap”, and add a polyadenylated tail. Luckily there are off-the-shelf algorithms like STAR that account for this variability. When aligned reads are part of a “transcriptomics” study, the reads are used to product an “expression matrix,” a [20,000 x 1] vector showing how much each gene is being transcribed:

When we combine transcriptomics data with SNP calls, we can do something called “expression quantitative trait loci” (eQTL) analysis, where we infer how a given SNP affects the transcript levels of nearby genes.
Structural variant calling
SNPs aren’t the only way one genome can differ from another. Genomes can also undergo “structural variation,” usually defined as a deletion, insertion, or mutation of >50bp or <1 million bp.
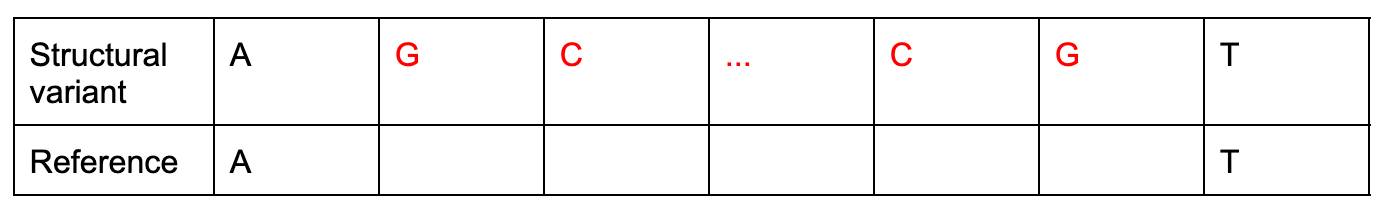
Detecting structural variants is challenging for the same reasons that counting RNA transcripts is hard: individual reads may have very poor match to the reference genome due to insertions or deletions. Once a researcher successfully detects a structural variant, they can perform an analysis very similar to eQTL in order to understand the impact of that variant on gene expression.
While I am not an expert in the Iran Hostage Crisis, I do love movies, and I can assert that only the Iranian’s successful alignment of those documents was able to set the stage for Argo’s suspenseful tarmac climax. Successful read alignment serves much the same role in bioinformatics by enabling sensational downstream analyses. Unfortunately read alignment can be challenging to do at scale, but that’s a discussion for a second post.
> But of course the reason we bother sequencing genomes is that they are not an exact match with the reference genome.
I always thought even getting the original reference genome was hard because reads could come from multiple points in the genome. Is that true? e.g.
genome:
AATAA
reads:
AAT
TAA
possible other genomes:
TAAT
AATAAT
although I guess this problem should be easier as you get more and/or longer reads.